
A modular tool to aggregate results from bioinformatics analyses across many samples into a single report.
This report has been generated by the nf-core/checkatlas analysis pipeline. For information about how to interpret these results, please see the documentation.
Report
generated on 2023-07-22, 02:21
based on data in:
/data/analysis/data_becavin/checkatlas_test/tuto/work/65/9ba8bb82797022b592961641925d3d
Checkatlas Qc Report



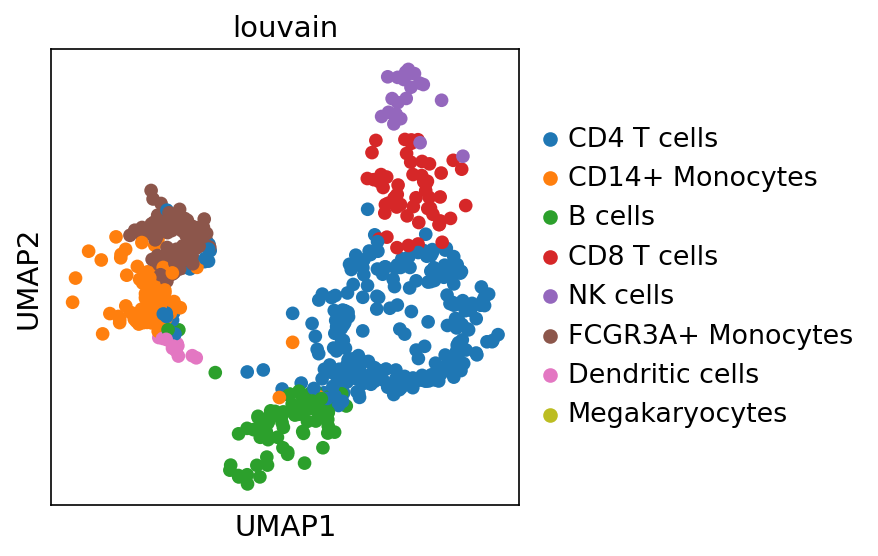
Checkatlas Umap Report

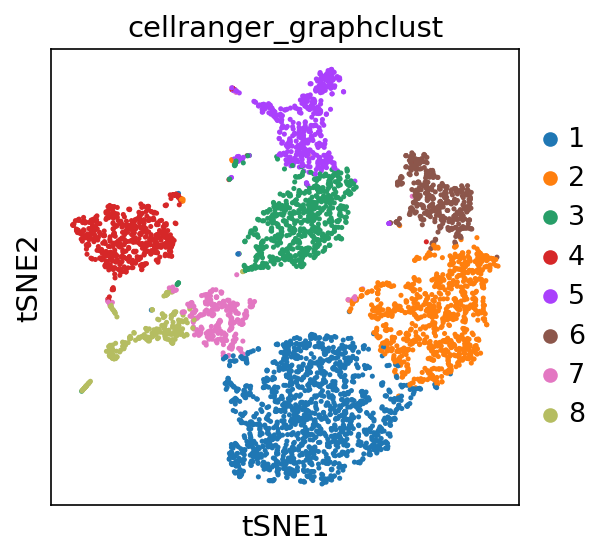
Checkatlas Tsne Report
CheckAtlas
CheckAtlas A one-liner tool for quality control of your single-cell atlases.
Atlas overview
Overview of your single-cell atlases
| Sample Name | AtlasFileType | NbCells | NbGenes | AnnData.raw | AnnData.X | File_extension | File_path |
|---|---|---|---|---|---|---|---|
| pbmc_3k_68k_integrated | AnnData | 700.0 | 208.0 | True | True | pbmc_3k_68k_integrated | /data/analysis/data_becavin/checkatlas_test/tuto/data4/pbmc_3k_68k_integrated.h5ad |
| pbmc_3k_68k_integrated_checkatlas_summ | Scanpy | 700.0 | 208.0 | True | True | .h5ad | /pbmc_3k_68k_integrated.h5ad |
| pbmc_3k_v1 | Cellranger < v3 | 2700.0 | 32738.0 | False | True | pbmc_3k_v1 | /data/analysis/data_becavin/checkatlas_test/tuto/data4/pbmc_3k_v1/outs/filtered_gene_bc_matrices/hg19/matrix.mtx |
| pbmc_68k_v1 | Cellranger < v3 | 68579.0 | 32738.0 | False | True | pbmc_68k_v1 | /data/analysis/data_becavin/checkatlas_test/tuto/data4/pbmc_68k_v1/outs/filtered_matrices_mex/hg19/matrix.mtx |
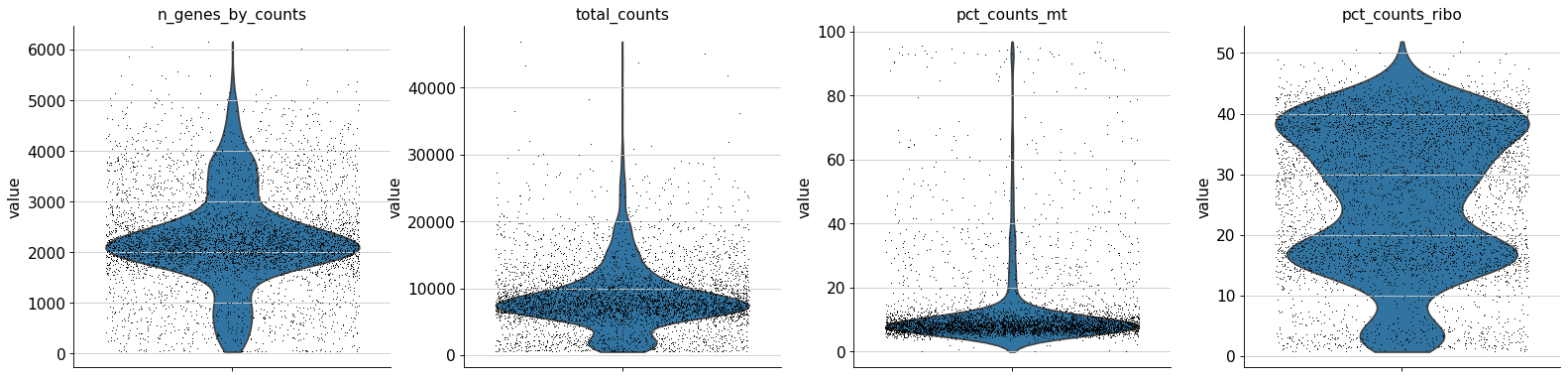
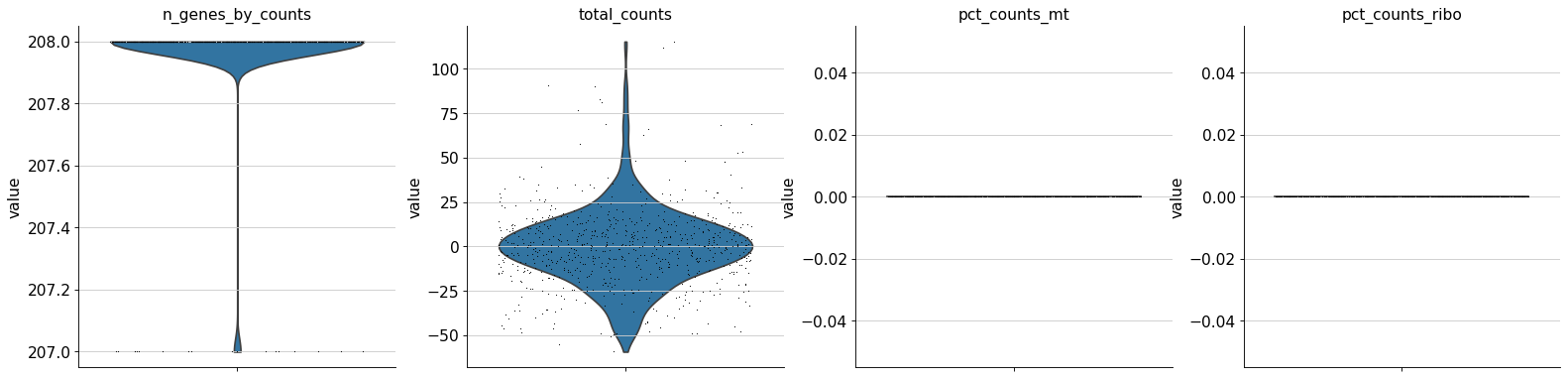
QC total_counts
QC of your atlases log10(Cellrank vs total-counts.
QC n_genes_by_counts
QC of your atlases log10(Cellrank vs n_genes_by_counts.
QC pct_counts_mt
QC of your atlases log10(Cellrank vs pct_counts_mt.
Classification metrics
Quality control metrics calculated on your atlases.
| Sample Name | obs | davies_bouldin |
|---|---|---|
| pbmc_3k_68k_integrated_louvain | louvain | 0.8 |
| pbmc_3k_v1_cellranger_kmeans_10 | cellranger_kmeans_10 | 1.7 |
Dimensionality reduction metrics
Quality control metrics calculated on your atlases.
| Sample Name | obsm |
|---|---|
| pbmc_3k_68k_integrated_X_pca | X_pca |
| pbmc_3k_68k_integrated_X_umap | X_umap |
| pbmc_3k_68k_integrated_rep | rep |
Atlas object explorer
Exploration of your Atlas object (Scanpy, Cellanger, Seurat)
| Sample Name | atlas_obs | obsm | var | varm | uns |
|---|---|---|---|---|---|
| pbmc_3k_68k_integrated | bulk_labelsn_genespercent_miton_countsS_scoreG2M_scorephaselouvain | X_pcaX_umaprep | n_countsmeansdispersionsdispersions_normhighly_variable | PCs | bulk_labels_colorslouvainlouvain_colorsneighborspcarank_genes_groups |
| pbmc_3k_v1 | cellranger_kmeans_10 | | gene_ids | | |
| pbmc_68k_v1 | cellranger_kmeans_10 | | gene_ids | | |
nf-core/checkatlas Methods Description
Suggested text and references to use when describing pipeline usage within the methods section of a publication.
Methods
Data was processed using nf-core/checkatlas v1.0dev of the nf-core collection of workflows (Ewels et al., 2020), utilising reproducible software environments from the Bioconda (Grüning et al., 2018) and Biocontainers (da Veiga Leprevost et al., 2017) projects.
The pipeline was executed with Nextflow v23.04.1 (Di Tommaso et al., 2017) with the following command:
nextflow run nf-core-checkatlas -r dev --path /data/analysis/data_becavin/checkatlas_test/tuto/data4/References
- Di Tommaso, P., Chatzou, M., Floden, E. W., Barja, P. P., Palumbo, E., & Notredame, C. (2017). Nextflow enables reproducible computational workflows. Nature Biotechnology, 35(4), 316-319. doi: 10.1038/nbt.3820
- Ewels, P. A., Peltzer, A., Fillinger, S., Patel, H., Alneberg, J., Wilm, A., Garcia, M. U., Di Tommaso, P., & Nahnsen, S. (2020). The nf-core framework for community-curated bioinformatics pipelines. Nature Biotechnology, 38(3), 276-278. doi: 10.1038/s41587-020-0439-x
- Grüning, B., Dale, R., Sjödin, A., Chapman, B. A., Rowe, J., Tomkins-Tinch, C. H., Valieris, R., Köster, J., & Bioconda Team. (2018). Bioconda: sustainable and comprehensive software distribution for the life sciences. Nature Methods, 15(7), 475–476. doi: 10.1038/s41592-018-0046-7
- da Veiga Leprevost, F., Grüning, B. A., Alves Aflitos, S., Röst, H. L., Uszkoreit, J., Barsnes, H., Vaudel, M., Moreno, P., Gatto, L., Weber, J., Bai, M., Jimenez, R. C., Sachsenberg, T., Pfeuffer, J., Vera Alvarez, R., Griss, J., Nesvizhskii, A. I., & Perez-Riverol, Y. (2017). BioContainers: an open-source and community-driven framework for software standardization. Bioinformatics (Oxford, England), 33(16), 2580–2582. doi: 10.1093/bioinformatics/btx192
Notes:
- If available, make sure to update the text to include the Zenodo DOI of version of the pipeline used.
- The command above does not include parameters contained in any configs or profiles that may have been used. Ensure the config file is also uploaded with your publication!
- You should also cite all software used within this run. Check the "Software Versions" of this report to get version information.
nf-core/checkatlas Software Versions
are collected at run time from the software output.
| Process Name | Software | Version |
|---|---|---|
| CUSTOM_DUMPSOFTWAREVERSIONS | python | 3.10.12 |
| yaml | 6.0.1 | |
| LIST_CELLRANGER_ATLASES | checkatlas | 0.3.9 |
| LIST_SCANPY_ATLASES | checkatlas | 0.3.9 |
| LIST_SEURAT_ATLASES | checkatlas | 0.3.9 |
| Workflow | Nextflow | 23.04.1 |
| nf-core/checkatlas | 1.0dev |
nf-core/checkatlas Workflow Summary
- this information is collected when the pipeline is started.
Core Nextflow options
- revision
- dev
- runName
- condescending_yonath
- launchDir
- /data/analysis/data_becavin/checkatlas_test/tuto
- workDir
- /data/analysis/data_becavin/checkatlas_test/tuto/work
- projectDir
- /home/becavin/.nextflow/assets/becavin-lab/nf-core-checkatlas
- userName
- becavin
- profile
- standard
- configFiles
- N/A
Input/output options
- path
- /data/analysis/data_becavin/checkatlas_test/tuto/data4/
- outdir
- N/A
